

Molegro Virtual Docker 3.0.0
Molegro Virtual Docker 3.0.0 | 15,9 MB
Molegro Virtual Docker is an integrated platform for predicting protein - ligand interactions. Molegro Virtual Docker handles all aspects of the docking process from preparation of the molecules to determination of the potential binding sites of the target protein, and prediction of the binding modes of the ligands. Molegro Virtual Docker provides the user with high-quality docking based on a novel optimization technique combined with a user interface experience focusing on productivity and usability. The Molegro Virtual Docker (MVD) has been shown to yield higher docking accuracy than other state-of-the-art docking products (MVD: 87%, Glide: 82%, Surflex: 75%, FlexX: 58%).
Here are some key features of "Molegro Virtual Docker":
Molecule Import and Preparation:
· It is easy to import and prepare molecules.
· Molecular structure files are parsed into relevant components (ligands, cofactors, water molecules and proteins) and can be automatically prepared.
· Parsing warnings and errors are shown in a convenient hierarchy.
· The built-in cavity detector identifies promising binding locations, making it possible to restrict the search space to the most interesting regions.
· A protonation wizard allows you to quickly identify potential protonation targets (e.g. histidine residues) and change their protonation state quickly using a context menu.
Docking:
The Docking process is managed via the Docking Wizard. The wizard allows you to:
· Select which structures to include in the simulation.
· Choose the potential binding region (MVD will automatically display potential binding pockets).
· Configure search algorithm properties and set up clustering and data logging.
· Manage additional constraints.
· Inspect warnings about unlikely preparations and missing structural information (e.g. unknown residues).
Analysis:
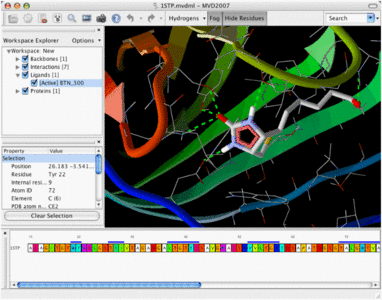
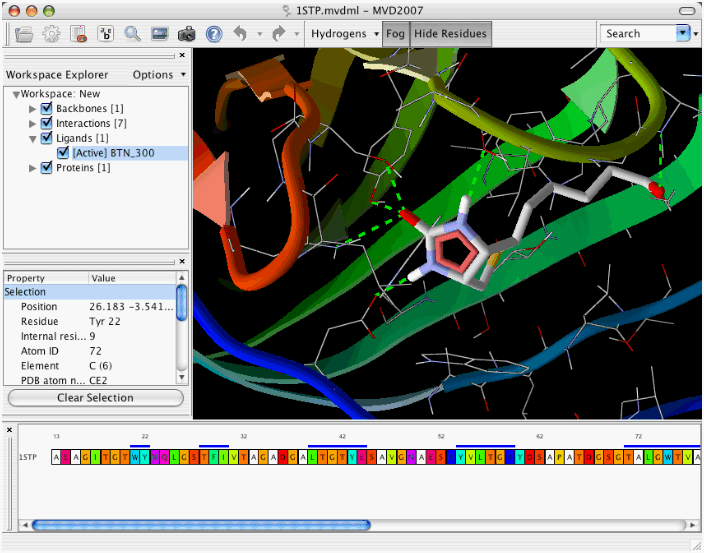
· The Pose Organizer allows you to browse the poses returned by the docking engine. It is capable of loading poses from a docking run dynamically, making it possible to browse thousands of ligands.
· It is possible to inspect the various energy terms and interactions and it is also possible to calculate the more advanced reranking and binding affinity measures. Hydrogen bonds and electrostatic interactions are updated dynamically when switching between poses.
Sequence and Structure Visualization:
· The Sequence Viewer allows you to quickly identify and select residues in a protein.
· The cartoon visualization highlights the different regions of secondary structure.
Constraints:
· Various constraints makes it possible to alter the energy landscape (e.g. by rewarding certain regions of the search space) or to force or prevent certain interactions from occurring.
· Constraints can be defined based on chemical properties (e.g. hydrogen bond acceptors or ring atoms) or individual atoms can be specified for each ligand.
Data Analyzer:
· The Data Analyzer in MVD can create regression models (either neural networks or multiple linear regression), extract statistical measures, and visualize data.
· Predict numerical properties of docked compounds from MVD docking runs. User-created models can be applied directly in the Pose Organizer.
· Create regression models using imported numerical descriptors (e.g. from third-party software).
· Feature selection for finding the relevant descriptors.
· Algebraic data transformation plugin for manipulating descriptors or creating derived descriptors.
· 1D plots (histograms), 2D plots (X-Y plots), and 3D plots.
· Various statistical measures.
Sidechain Flexibility:
· In MVD, sidechain flexibility is implemented by softening the potential during the docking (by increasing the tolerance of the PLP-potentials or by weakening selected sidechains interactions), docking a diverse set of poses, and finally optimizing the sidechain configurations. It is also possible to manually minimize a receptor structure.
· After the docking simulation has completed, the found poses can be inspected simultaneously with their matching receptor conformation directly in the pose organizer.
Template Docking:
MVD makes it possible to dock ligands against docking templates. Similar to pharmacophores, templates are build from relevant chemical properties (like charge and hydrogen bonding capabilities) and can be auto-generated from one or more ligand conformations. Templates can be used in several ways:
· To flexibly align a number of ligands (and determine a score for their similarity).
· To focus or guide the docking simulation by combining the template similarity score with a receptor-based docking scoring function.
· To extract a fine-grained similarity score from each template point and create a regression model using the Data Analyzer (3D QSAR).
Other Features:
· Runs on Mac (PowerPC and Intel), Linux (32-bit and 64-bit), and Windows machines.
· Extensible and customizable macro system with built-in editor.
· Customizable molecular surfaces and backbone representation.
· Biomolecule generator for PDB files containing symmetry transformation information.
· Advanced text labelling for various molecular properties.
· Online help and automatic check for updates.
· MVD can be scripted using either its own scripting language or by external scripting languages (a Python wrapper is included).
· Pose Modifier for manually modifying poses.